
Research Bio
Stephen R. Leone is a professor in the Department of Chemistry and the Department of Physics. He is the John R. Thomas Endowed Chair in Physical Chemistry and a faculty investigator of Lawrence Berkeley National Laboratory.
Professor Leone's research interests include ultrafast laser investigations in the soft X-ray and extreme ultraviolet (XUV) to probe valence and core levels, attosecond physics and chemistry, attosecond wave mixing and multidimensional spectroscopy, state-resolved decay processes and time dynamics investigations, in atoms, molecules, and solids
Current projects are grouped along a few main themes:
(1) ultrafast laser-induced molecular dynamics, including molecular geometry relaxation and dissociation reactions, electronic-nuclear dynamics across curve crossings and conical intersections, and formation and propagation of coherent superpositions;
(2) dynamics of photoexcited solids, including semiconductors, ferromagnetic metals, layered heterostructures, two-dimensional van der Waals materials, and nanoparticles, such as ultrafast charge and spin dynamics in thin films and nanostructures, and electron-lattice coupling in solar relevant semiconductors;
(3) development of new methods such as four wave mixing and multidimensional XUV spectroscopy.
Examples are: Ultrafast lasers are used to probe the coherent dynamics of molecular motion on the timescales of vibrational, rotational, or electronic periods. The study of molecular photodissociation by soft X-ray laser techniques has opened the way to analyze the simple breaking of a molecular bond in greater detail. High order harmonics in the XUV and soft X-ray are produced by strong laser fields in a rare gas, and used to probe transitions to valence orbitals by core level spectroscopy of time-evolving systems ranging from atoms to small molecules.
In particular, X-ray and XUV transient absorption spectroscopy is used to probe electronic superpositions, curve crossings, passage through conical intersections and molecular fragmentation pathways. By using few-cycle carrier-envelope phase-stabilized laser pulses, isolated attosecond pulses are generated to study electronic timescales in solids, thin films, molecules and nanoparticles.
A new, XUV magnetic circular dichroism (XMCD) apparatus is built to study element-specific, spin-resolved coherent dynamics in solid state systems, measurements that are valuable for quantum information systems that rely on spin transfer to exchange information. Core-level transient absorption spectroscopy of thin semiconductor and metal films reveals the few-femtosecond dynamics of electronic thermalization, and the simultaneous sub-picosecond electronic cooling and lattice heating via electron-phonon scattering, in a single measurement.
Attosecond wave mixing and multidimensional spectroscopy experiments are designed to elucidate ultrashort time processes of associated electronic excited states and core level transitions by extending highly selective nonlinear wave-mixing techniques developed in infrared, optical and radiofrequency spectroscopies to the XUV regime. These have extended the spectral window to the carbon K-edge in several transient absorption setups. A four-wave mixing setup aiming at C K-edge is also under development. The ability to directly probe carbon atoms is opening new avenues of ultrafast dynamics in various organic and bio-relevant molecules.
Recent Highlights
1. “Femtosecond symmetry breaking and coherent relaxation of methane cations via x-ray spectroscopy” Science 380, 713-717 (2023)
By establishing abrupt ionization using ultrashort visible-infrared pulses, the ultrafast geometrical relaxation of methane cations was probed via soft X-ray transient absorption spectroscopy at the C K-edge. The measurements reveal how methane cations undergo Jahn-Teller distortion in less than 10 fs and follow with an internal vibrational energy redistribution within 58 fs. Theory calculations back up the experimental results and link the observables to the change in the smallest H-C-H angle.
2. Nonlinear & noncollinear wave mixing spectroscopy experiments in the solid state
Attosecond noncollinear wave mixing spectroscopy experiments in the solid state were undertaken to understand the formation, behavior, interaction and decay on an ultrafast scale of various charge carriers and quasiparticles in insulators, semiconductors and metals. Attosecond XUV-NIR four-wave mixing (4WM) spectroscopy investigations in insulators such as NaCl obtained new results on the coherence decays of core excitons ["Solid state core-exciton dynamics in NaCl observed by tabletop attosecond four-wave mixing spectroscopy," Phys. Rev. B 103, 245140 (2021)]. Core excitons are short lived quasi-particles based on the attraction of electrons for inner shell holes. A similar methodology was recently successful to establish a transient grating in tellurium and to scatter XUV photons from the carrier grating.
Research Expertise and Interest
attosecond physics, attosecond chemistry, ultrafast spectroscopy, ultrafast laser, atomic and molecular dynamics, semiconductors, quantum materials, high harmonic generation, soft x-ray, charge transfer, charge migration, nonadiabatic dynamics, conical intersection, carrier dynamics, phonon dynamics, spin dynamics, core exciton dynamics
In the News

Watching Molecules Relax in Real Time
What happens when you explode a chemical bond?
Making molecular movies with X-rays

Scientists measure speedy electrons in silicon
In semiconductors like silicon, electrons attached to atoms in the crystal lattice can be mobilized into the conduction band by light or voltage. Berkeley scientists have taken snapshots of this very brief band-gap jump and timed it at 450 attoseconds.

For the first time ever, scientists watch an atom’s electrons moving in real time
A team of German and UC Berkeley scientists, including physicist Stephen Leone, have used ultrashort flashes of laser light to directly observe for the first time the movement of an atom’s outer electrons. The technique could be valuable in the study of photosynthesis.
Featured in the Media
By establishing abrupt ionization using ultrashort visible-infrared pulses, the ultrafast geometrical relaxation of methane cations was probed via soft X-ray transient absorption spectroscopy at the C K-edge. The measurements reveal how methane cations undergo Jahn-Teller distortion in less than 10 fs, create a coherent vibrational motion in the scissoring vibration, and follow with an internal vibrational energy redistribution within 58 fs. Theory calculations back up the experimental results and link the observables to the change in the smallest H-C-H angle.
In the Bohr model of the atom, the nominal orbiting period for a 1s electron in the hydrogen atom is 150 as (1 as = 10−18 s). Attosecond light pulses are thus in principle short enough to follow electron dynamics in real time (1). Moreover, with such pulses, the time-resolved measurements can be extended to subfemtosecond lifetimes, probe electronic and vibrational coherences in atoms and molecules, and by means of attosecond photochemistry, follow the earliest dynamics of photoexcited molecules as they dissociate or isomerize. Attosecond light pulses have recently been incorporated into four-wave mixing (FWM) experiments in gases and solids, by combining one attosecond pulse with two visible optical pulses or multiple attosecond pulses themselves. This now allows more accurate determination of quantum pathways in transient electronic states.
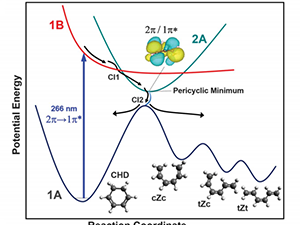
Electronic relaxation in organic chromophores often proceeds via states not directly accessible by photoexcitation. We report on the photoinduced dynamics of pyrazine that involves such states, excited by a 267 nm laser and probed with X-ray transient absorption spectroscopy in a table-top setup. In addition to the previously characterized 1B2u (ππ*) (S2) and 1B3u (nπ*) (S1) states, the participation of the optically dark 1Au (nπ*) state is assigned by a combination of experimental X-ray core-to-valence spectroscopy, electronic structure calculations, nonadiabatic dynamics simulations, and X-ray spectral computations. Despite 1Au (nπ*) and 1B3u (nπ*) states having similar energies at relaxed geometry, their X-ray absorption spectra differ largely in transition energy and oscillator strength. The 1Au (nπ*) state is populated in 200 ± 50 femtoseconds after electronic excitation and plays a key role in the relaxation of pyrazine to the ground state.
Attosecond noncollinear wave mixing spectroscopy experiments in the solid state were undertaken to understand the formation, behavior, interaction and decay on an ultrafast scale of various charge carriers and quasiparticles in insulators, semiconductors and metals. Attosecond XUV-NIR four-wave mixing (4WM) spectroscopy investigations in insulators such as NaCl obtained new results on the coherence decays of core excitons. Core excitons are short lived quasi-particles based on the attraction of electrons for inner shell holes. A similar methodology was recently successful to establish a transient grating in tellurium and to scatter XUV photons from the carrier grating.
The fragmentation of photoexcited iso-propyl iodide and tert-butyl iodide molecules (i-C3H7I and t-C4H9I) through a conical intersection between 3Q0/1Q1 spin–orbit states is revealed by ultrafast XUV transient absorption measuring iodine 4d core-to-valence transitions. The electronic state-sensitivity of the technique allows for a complete mapping of molecular dissociation from photoexcitation to photoproducts. In both molecules, the sub-100 fs transfer of a photoexcited wave packet from the 3Q0 state into the 1Q1 state at the conical intersection is captured. The results show how differences in the electronic state-switching of the wave packet in i-C3H7I and t-C4H9I directly lead to differences in the photoproduct branching ratio of the two systems.
Charge transport and recombination kinetics in each layer of a Ni-TiO2-Si junction is measured using the element specificity of broadband extreme ultraviolet (XUV) ultrafast pulses. After silicon photoexcitation, holes are inferred to transport from Si to Ni ballistically in ~100 fs, resulting in characteristic spectral shifts in the XUV edges. Meanwhile, the electrons remain on Si. After picoseconds, the transient hole population on Ni is observed to back-diffuse through the TiO2, shifting the Ti spectrum to a higher oxidation state, followed by electron-hole recombination at the Si-TiO2 interface and in the Si bulk. Electrical properties, such as the hole diffusion constant in TiO2 and the initial hole mobility in Si, are fit from these transient spectra and match well with values reported previously
Attosecond transient absorption spectroscopy is employed to follow the valence dynamics of strong-field initiated processes in methyl bromide. By probing the 3d core-to-valence transition, we resolve the strong field excitation and ensuing fragmentation of the neutral σ* excited states of methyl bromide. The results provide a clear signature of the non-adiabatic passage of the excited state wave packet through a conical intersection. We additionally observe competing, strong field initiated processes arising in both the ground state and ionized molecule corresponding to vibrational and spin-orbit motion, respectively. The demonstrated ability to resolve simultaneous dynamics with few-femtosecond resolution presents a clear path forward in the implementation of attosecond XUV spectroscopy as a general tool for probing competing and complex molecular phenomena with unmatched temporal resolution.
The electronic character of photoexcited molecules can abruptly change at avoided crossings and conical intersections. Here, we report direct mapping of the coupled interplay between electrons and nuclei in a prototype molecule, iodine monobromide (IBr), by using attosecond transient absorption spectroscopy. A few-femtosecond visible pulse resonantly excites the Y(0+), and Z(0+) states of IBr, and the photodissociation dynamics are tracked with an attosecond extreme-ultraviolet pulse that simultaneously probes the I-4d and Br-3d core-level absorption edges. Direct comparison with quantum mechanical simulations unambiguously identifies the absorption features associated with adiabatic and diabatic channels at the B/Y avoided crossing and concurrent two-photon dissociation processes that involve the Y/Z avoided crossing. The results show clear evidence for rapid switching of valence-electronic character at the avoided crossing.