Using CRISPR, new technique makes it easy to map genetic networks
CRISPR-Cas9 makes it easy to knock out or tweak a single gene to determine its effect on an organism or cell, or even another gene. But what if you could perform several thousand experiments at once, using CRISPR to tweak every gene in the genome individually and quickly see the impact of each?
A team of University of California, Berkeley, scientists has developed an easy way to do just that, allowing anyone to profile a cell, including human cells, and rapidly determine all the DNA sequences in the genome that regulate the expression of a specific gene.
While the technique will mostly benefit basic researchers who are interested in tracking the cascade of genetic activity — the genetic network — that impacts a gene they’re interested in, it will also help researchers quickly find the regulatory sequences that control disease genes and possibly find new targets for drugs.
“A disease where you might want to use this approach is cancer, where we know certain genes that those cancer cells express, and need to express, in order to survive and grow,” said Nicholas Ingolia, UC Berkeley associate professor of molecular and cell biology. “What this tool would let you do is ask the question: What are the upstream genes, what are the regulators that are controlling those genes that we know about?”
Those controllers may be easier to target therapeutically in order to shut down the cancer cells.
The new technique simplifies something that has been difficult to do until now: backtrack along genetic pathways in a cell to find these ultimate controllers.
“We have a lot of good ways of working forward,” he said. “This is a nice way of working backward, figuring out what is upstream of something. I think it has a lot of potential uses in disease research.”
“I sometimes use the analogy that when we walk into a dark room and flip a light switch, we can see what light gets switched on. That light is like a gene, and we can tell, when we flip the switch, what genes it turns on. We are already very good at that,” he added. “What this lets us do is work backward. If we have a light we care about, we want to find out what are the switches that control it. This gives us a way to do that.”
Ryan Muller, a graduate student in the Ingolia lab, and colleagues Lucas Ferguson and Zuriah Meacham, along with Ingolia, will publish the details of their technique online on Dec. 10 in the journal Science.
Barcoding the genome
Since the advent of CRISPR-Cas9 gene-editing eight years ago, researchers who want to determine the function of a specific gene have been able to precisely target it with the Cas9 protein and knock it out. Guided by a piece of guide RNA complementary to the DNA in the gene, the Cas9 protein binds to the gene and cuts or, as with CRISPR interference (CRISPRi), inhibits it.
In the crudest type of assay, the cell or organism either lives or dies. However, it’s possible to look for more subtle effects of the knockout, such as whether a specific gene is turned on or off, or how much it’s turned up or down.
Today, that requires adding a reporter gene — often one that codes for a green fluorescent protein — attached to an identical copy of the promoter that initiates expression of the gene you’re interested in. Since each gene’s unique promoter determines when that gene is expressed, if the Cas9 knockout affects expression of your gene of interest, it will also affect expression of the reporter, making the culture glow green under fluorescent light.
Nevertheless, with 6,000 total genes in yeast — and 20,000 total genes in humans — it’s a big undertaking to tweak each gene and discover the effect on a fluorescent reporter.
“CRISPR makes it easy to comprehensively survey all the genes in the genome and perturb them, but then the big question is, How do you read out the effects of each of those perturbations?” he said.
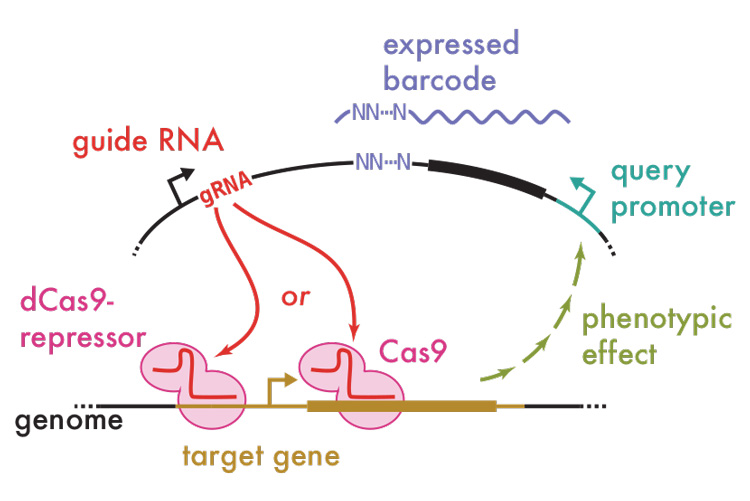
This new technique, which Ingolia calls CRISPR interference with barcoded expression reporter sequencing, or CiBER-seq, solves that problem, allowing these experiments to be done simultaneously by pooling tens of thousands of CRISPR experiments. The technique does away with the fluorescence and employs deep sequencing to directly measure the increased or decreased activity of genes in the pool. Deep sequencing uses high-throughput, long-read next generation sequencing technology to sequence and essentially count all the genes expressed in the pooled samples.
“In one pooled CiBER-seq experiment, in one day, we can find all the upstream regulators for several different target genes, whereas, if you were to use a fluorescence-based technique, each of those targets would take you multiple days of measurement time,” Ingolia said.
CRISPRing each gene in an organism in parallel is straightforward, thanks to companies that sell ready-made, single guide RNAs to use with the Cas9 protein. Researchers can order sgRNAs for every gene in the genome, and for each gene, a dozen different sgRNAs — most genes are strings of thousands of nucleotides, while sgRNAs are about 20 nucleotides long.
The team’s key innovation was to link each sgRNA with a unique, random nucleotide sequence — essentially, a barcode — connected to a promoter that will only transcribe the barcode if the gene of interest is also switched on. Each barcode reports on the effect of one sgRNA, individually targeting one gene out of a complex pool of thousands of sgRNAs. Deep sequencing tells you the relative abundances of every barcode in the sample — for yeast, some 60,000 — allowing you to quickly assess which of the 6,000 genes in yeast has an effect on the promoter and, thus, expression of the gene of interest. For human cells, a researcher might insert more than 200,000 different guide RNAs, targeting each gene multiple times.
“This is really the heart of what we were able to do differently: the idea that you have a big library of different guide RNAs, each of which is going to perturb a different gene, but it has the same query promoter on it — the response you are studying. That query promoter transcribes the random barcode that we link to each guide,” he said. “If there is a response you care about, you poke each different gene in the genome and see how the response changes.”
If you get one barcode that is 10 times more abundant than any of the others, for example, that tells you that that query promoter is switched on 10 times more strongly in that cell. In practice, Ingolia attached about four different barcodes to each guide RNA, as a quadruple check on the results.
“By looking more directly at a gene expression response, we can pick up on a lot of subtlety to the physiology itself, what is going on inside the cell,” he said.
In the newly reported experiments, the team queried five separate genes in yeast, including genes involved in metabolism, cell division and the cell’s response to stress. While it may be possible to study up to 100 genes simultaneously when CRISPRing the entire genome, he suspects that, for convenience, researchers would limit themselves to four or five at once.
The work was funded by the National Institutes of Health (DP2 CA195768, R01 GM130996).