Researcher tweaks cells with mRNA, in quest to improve antivirals

Herpesviruses are master manipulators. Once inside the body, they can turn healthy cells into virus factories within a few short hours.
“These tiny organisms come with few of their own resources, yet they’re exquisitely adept at taking control of their far more powerful host,” says Britt Glaunsinger. “That makes them fantastic teachers of our own biology.” A Berkeley professor of biochemistry and molecular biology, Glaunsinger studies the cell takeover tactics of gamma herpesviruses. Their interactions with human cells are illuminating how cells regulate their own gene expression via RNA.
Normally, a cell transcribes its genes into messenger RNA (mRNA) templates within the nucleus. These templates are then exported into the cell’s main compartment, the cytoplasm, and made into proteins.
In cells infected by gamma herpesviruses, Glaunsinger has found that a protein called SOX degrades or eliminates most cytoplasmic mRNA. It separates molecules called poly-A binding proteins, or PABPs, from mRNA, destabilizing the mRNA in the process. “That frees up the translation machinery to focus on viral genes—the virus doesn’t have to compete with the cell’s gene expression,” Glaunsinger says.
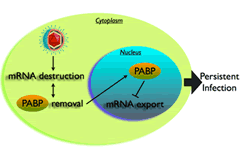
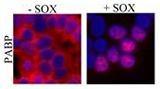
Her work has also shown that SOX shrinks the supply of mRNAs to a trickle. It boots all PABPs out of the cytoplasm and into the nucleus. There, PABP alters host mRNAs, making them appear damaged. The cell’s own proofreading system prevents these faulty transcripts from leaving the nucleus.
A number of different, unrelated viruses use PABP like SOX does—to restrict gene expression. When many viruses converge on the same pathway within a cell, Glaunsinger says, “that gives us a clue that a pathway is really important; it’s a master regulator of gene expression.”
Glaunsinger suspects that PABP relocation may actually be a normal cellular reaction to certain types of stress. By pausing gene expression, she says, stressed cells can assess the situation and potentially devote their energies elsewhere. Herpesviruses are merely taking advantage of that feature. “Viruses make do with what the cell has and figure out how to make that work for them,” Glaunsinger says.
Yet putting the brakes on gene expression seems like the last thing a virus would want to do. All germs are in a race to replicate before their host cell dies or gets killed by the immune system.
Glaunsinger’s most recent findings may help explain this paradox. She made a point mutation in the SOX gene that, in cultured cells, made the virus ineffective at blocking host gene expression. In mice, the virus also initially replicated without a problem. Unexpectedly however, the mutant proved unable to persist for longer periods of time in the mice. That meant it could not enter the second phase of its life cycle: the latent, or inactive, stage that herpes and other viruses use to linger in the body for decades.
Glaunsinger suspects viruses modulate host gene expression for two reasons: to keep the host cell alive, and to evade immune system eradication. “If you had really high viral gene expression, the cell could get overwhelmed and die before releasing infectious virions. And dampening a cell’s ability to express its own genes means it can’t make proteins that alert the immune system to its own infection,” she says.
Intrigued by these findings, Glaunsinger is now investigating what made the mutant virus so impotent. What she uncovers could help scientists flush viruses out of their inactive, latent phase and into their replicating, lytic phase. “Antivirals only work on viruses that are replicating,” Glaunsinger says. “Figuring out ways to lure latent viruses out of hiding en masse is critical for curing somebody” from shingles, herpes or HIV. Eradicating such viruses could also reduce cases of cancers, as viruses create microenvironments that promote tumors. But until such a treatment is found, tiny viruses will continue to exert an outsized influence upon us all.
This article originally appeared in Science Matters (vol. 7, issue 55)