Learning to Read the Genome
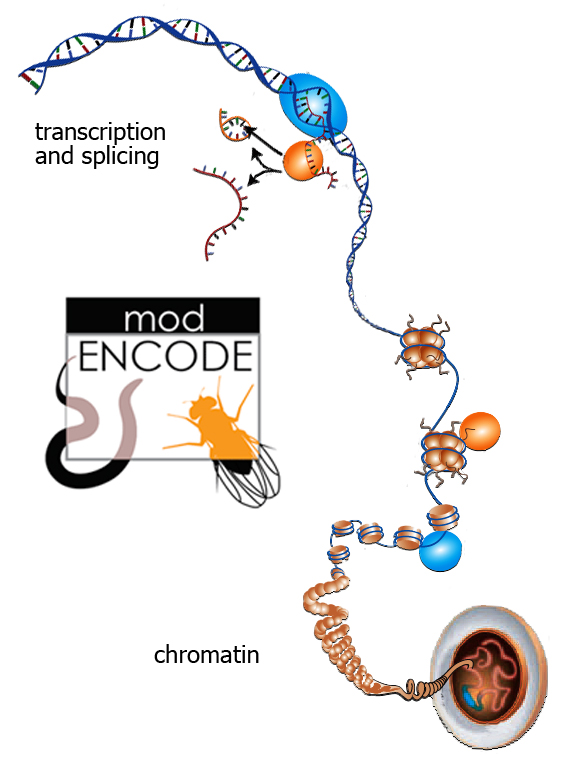
In the past decade researchers have made astonishing progress in the rapid and accurate sequencing of genomes from all realms of life. Yet the listing of chemical base pairs has gotten far ahead of understanding how the information they contain becomes functional. Even the best-understood genomes conceal mysteries.
Genetic information carried by DNA and RNA operates together with the patterns and physical organization of chromosomes to produce a working organism. Major advances in understanding these complex relationships are published this week by the “model organism Encyclopedia of DNA Elements” (modENCODE) project, funded by the National Institutes of Health’s National Human Genome Research Institute. These new insights into reading the genome apply not only to the fruit fly Drosophila melanogaster and the roundworm Caenorhabditis elegans, modENCODE’s two model organisms, but will apply to human beings and many other organisms as well.
Susan Celniker and Gary Karpen of the Life Sciences Division at the U.S. Department of Energy’s Lawrence Berkeley National Laboratory lead two of the principal research groups in the Drosophila modENCODE Consortium. They are among the senior co-authors of the Consortium’s report on integrating Drosophila functional elements and regulatory circuits, led by Manolis Kellis of the Massachusetts Institute of Technology, which appears in the December 24 issue of Science now online. Separate papers by the Celniker- and Karpen-led groups will appear in Nature in January and are now available online, and more papers by their groups will soon appear in an issue of Genome Research devoted to modENCODE studies.
The landscape of the genome
“Drosophila may be the single most thoroughly studied model organism; it allowed us to discover, a century ago, that chromosomes are the carriers of genetic information,” says Celniker, whose group studies the transcriptome. The transcriptome is the totality of RNA forms that transmit genetic information to the cellular machinery which constructs functioning proteins, as well as the noncoding RNAs that regulate gene expression, splicing, RNA stability, and metabolism. Yet, says Celniker, “there’s still a lot of undiscovered territory.”
Celniker’s transcriptome group succeeded in exploring Drosophila RNAs at a level never achieved before. “From the RNAs we identified approximately one thousand new genes, both protein-coding and noncoding. These were previously missed because they are less well conserved, or were found in less-studied developmental stages and RNA populations. Thus they tend to be expressed at lower levels than known genes.” She adds, “We also found an order-of-magnitude increase in the ways that genes are spliced and edited to produce alternate forms of known proteins, thus significantly increasing the complexity of the proteome.” The proteome is the set of all proteins expressed by the genome.
Karpen’s group studies chromatin, the combination of DNA and proteins that organize an organism’s genome into chromosomes. In chromatin, the DNA is wound around structures called nucleosomes, made of histone proteins. Other proteins (and some RNA) in chromatin also affect its organization and function. Karpen says the group’s goal “is to define the distributions of chromatin proteins and how chemical modifications can change their function.” They have produced the first comprehensive picture of how patterns of chromatin components are associated with chromosome functions, including the active transcription of genes. These mechanisms are called “epigenetic” because their influence on genome function is coded by the associated proteins rather than the DNA sequence.
The modENCODE transcriptome and chromatin groups, working with other groups that concentrate on regulatory elements, small RNAs, and DNA replication, produced what Karpen calls a “groundbreaking, comprehensive analysis, which vastly increases the information about the Drosophila genome available to researchers and provides a foundation for in-depth functional studies.”
The research groups carried out their studies on four different kinds of Drosophila cells maintained in laboratory cell cultures, not all of which had been extensively explored before. Additional studies with whole animals were carried out, especially in tracking developmental changes, from fly embryos through larvae and pupae to adult males and females.
Exploring the transcriptome, illuminating chromatin
Using a variety of techniques, the researchers developed 700 new data sets of information on different aspects of the fly genome. The transcriptome group identified 17,000 genes, both coding and noncoding, of which 1,938 were new.
But DNA is surprisingly versatile – coding sequences, known as exons, can be spliced together in different ways to produce more than one form of a protein. The researchers found almost 53,000 new or modified exons and almost 23,000 new splicing junctions, with 14,000 alternative ways of transcribing the genetic information. Despite the scrutiny to which the Drosophila genome has been subjected, the researchers found new or altered exons or splice forms in almost three-quarters of Drosophila’s previously annotated genes.
Like all eukaryotes (organisms whose cell nuclei are enclosed within a membrane) Drosophila’s genome is divided among euchromatin, which contains many active genes, and heterochromatin, which – although it amounts to about a third of the genome – contains relatively few active genes. Thus the Drosophila chromatin group was surprised to discover that some regions of heterochromatin are almost as active in expression as euchromatin.

The mark of an active or silent chromatin region is the chemical state of its nucleosomes, specifically whether the histones, on which the DNA is wrapped, permit or prevent the RNA-constructing enzyme, RNA polymerase, to bind to the DNA for transcription. For example, acetylated histones generally promote transcription, while many methylated histones can repress transcription. The Drosophila chromatin group found that in some regions, what controlled gene expression could not be identified from the DNA sequence, yet these regions were marked by specific histone modifications and other epigenetic factors. They also found active regions of euchromatin that carried marks characteristic of heterochromatin, patterns that were a combination of both “active” and “silent” marks.
By identifying the combinatorial patterns of 18 different histone modifications, and analyzing their associations with gene expression and other functions, the group developed a model of chromatin states working in concert, and how these vary among different cell lines. Their model identified novel chromatin signatures associated with regulation of gene activity and other functions, as well as many previously unidentified genes and promoters.
Of the modENCODE project, Celniker says, “The goal is not only to map every base in the genome but to discover the function of every base.” Adds Karpen, “Discovering function starts with mapping all the components that affect it.”
The promise of modENCODE
The results of the modENCODE research go into a central, publicly accessible database. Karpen says, “This information is available for any scientist to use in designing and conducting his or her own experiments. They can use our data to interrogate their favorite genes or the entire genome. It’s from their creativity and ingenuity that progress in understanding the flow of information from sequence to cell and organismal functions will be made.”
From the genomes of model organisms like the fruit fly and the roundworm, says Celniker, “We would like to crack the genomic code and discover the rules required to read a genome – any genome. Knowing which signals control gene expression in the fruit fly and the roundworm, including how chromatin affects gene expression, will be applicable to understanding how to read the human genome.”
“Identification of Functional Elements and Regulatory Circuits by Drosophila modENCODE,” by the Drosophila modENCODE Consortium, appears in the 24 December 2010 issue of Science and is now online. The Consortium’s transcriptome group, led by Berkeley Lab’s Susan Celniker, includes research teams led by Brenton Graveley of the University of Connecticut Health Center, Peter Cherbas of Indiana University Bloomington, Tom Gingeras of Cold Spring Harbor Laboratory, Norbert Perrimon of Harvard Medical School, Michael Brent of Washington University in Saint Louis, and Steven Brenner of the University of California at Berkeley. The chromatin group, led by Gary Karpen of Berkeley Lab and UC Berkeley, includes teams led by Sarah Elgin of Washington University in Saint Louis, Mitzi Kuroda of Harvard Medical School, Peter Park of Harvard Medical School, and Vince Pirrotta of Rutgers University.
“The developmental transcriptome of Drosophila melanogaster,” by members of the transcriptome group, and “Comprehensive analysis of the chromatin landscape in Drosophila melanogaster,” by members of the chromatin group, now appears in advance online publication of Nature.
Forthcoming papers on promoter architecture, by Celniker and first author Roger Hoskins of her team at Berkeley Lab; on transcriptional profiling of Drosophila cell lines, by Lucy and Peter Cherbas of the Celniker group; and on chromatin organization of heterochromatin by the Karpen group, including co-first author Aki Minoda of Berkeley Lab, will appear in a forthcoming issue of Genome Research devoted to modENCODE studies.