DNA repair after CRISPR cutting not at all what people thought
Despite high hopes and high investment in CRISPR-Cas9 gene editing, scientists still have a lot to learn about how it works in humans.
In the latest example, University of California, Berkeley, scientists found that people’s assumptions about how cells repair the genome after the Cas9 enzyme snips DNA are wrong.
The discovery gives insight into why CRISPR-Cas9 gene editing works remarkably well in nearly every cell attempted, though not with equal success in all cells. And it could help researchers boost the efficiency with which cells insert new DNA into the genome – to replace a harmful mutation with the correct DNA sequence, for example – and generally tweak CRISPR-Cas9 editing to get the desired outcome.
“If you want to treat sickle cell anemia, your chances of success are inextricably tied to the efficiency with which you can replace the mutated sickle cell gene with the correct one,” said UC Berkeley postdoctoral fellow Chris Richardson, first author of a paper describing the findings. “If you harvest a million cells from a patient and you have 10 percent insertion rate, that is not as good as if you have 30 to 40 percent. Being able to manipulate those cells to increase the frequency of this process, called homology-directed repair, is exciting.”
“Gene editing is super-powerful, with a lot of promise, but, so far, a lot of trial and error. The way it works in human cells has been a black box with a lot of assumptions,” said lead author Jacob Corn, a UC Berkeley adjunct professor of molecular and cell biology. “We are finally starting to get a picture of what’s going on.”
Corn, Richardson and their colleagues will publish their findings in the August issue of the journal Nature Genetics, available online now.
Corn was until recently the scientific director of biomedicine in the Innovative Genomics Institute, a joint CRISPR research program between UC Berkeley and UC San Francisco. This fall, he will join the faculty of ETH in Zurich, Switzerland.
CRISPR relies on DNA repair
CRISPR-Cas9 is revolutionary because of the precision with which it homes in on a specific DNA sequence out of billions in the genome and cleaves the double-stranded DNA molecule. But after that, it’s up to the cell to repair the damage.
Repair can happen in two ways. Enzymes can stitch the dangling ends back together, which often results in one or more bases – the building blocks of DNA – being added or deleted, disrupting the function of the gene. Alternatively, other enzymes can patch the break with a single strand of DNA that matches the DNA sequence upstream and downstream of the cut. A complementary DNA strand is created to complete the double-strand repair.
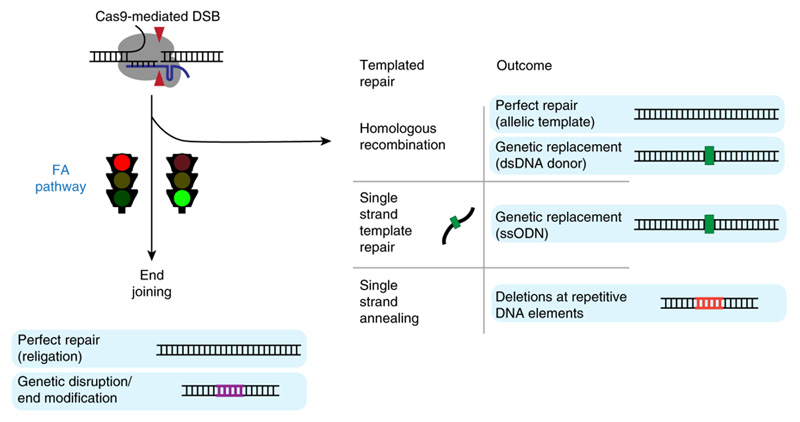
The former, called non-homologous end-joining, appears to be the most common outcome after CRISPR cutting. The latter, homology-directed repair, happens more frequently in some types of cells than others, and requires the presence of a piece of DNA that can be used to patch the break. Researchers often supply a single-stranded piece of DNA and hope that the cell uses it to replace the faulty sequence with the new one.
Both processes are a bit mysterious, however, and no one knows why some cells readily patch in DNA while others do so infrequently.
“The enthusiasm for using CRISPR-Cas9 for medical or synthetic biology applications is great, but no one really knows what happens after you put it into cells,” Richardson said. “It goes and creates these breaks and you count on the cells to fix them. But people don’t really understand how that process works.”
To find out which DNA repair enzymes are critical to homology-directed repair after CRISPR cutting, Richardson and Corn employed a technique called CRISPR interference (CRISPRi) to knock out, one at a time, more than 2,000 genes known or suspected to be involved in DNA repair, a function critical to a healthy cell.
Surprisingly, many of the genes that proved to be important – homology-directed repair dropped dramatically when they were silenced – were involved in an important repair system not thought to be involved in CRISPR repair.
Fanconi anemia
The pathway involves 21 separate proteins and is called the Fanconi anemia pathway because, if any of the genes for these proteins is damaged, people develop Fanconi anemia, a rare but serious hereditary disease in which the bone marrow cannot make enough new blood cells. It is associated with birth defects and a high risk of cancer, including a 10 percent chance of developing leukemia in childhood. Few patients live beyond 30 years of age.
The pathway has been known and studied for decades, but it was largely understood to repair one specific kind of DNA damage: DNA interstrand crosslinks, where a nucleotide on one strand of DNA bonds tightly with a nucleotide on the adjacent strand, interfering with DNA replication and often killing the cell. Researchers in the 1980s had reported a connection between homology-directed repair and the Fanconi anemia pathway, but it had been ignored or misunderstood, Corn noted.
“Based on our work, we believe that the Fanconi anemia pathway plays a major role in fixing other types of lesions as well, but is best understood as the pathway that repairs double-strand breaks,” Richardson said. “After Cas9 editing, the Fanconi anemia pathway is required if you want to insert new DNA.”
The importance of the Fanconi anemia pathway in repairing CRISPR breaks throws into doubt some planned CRISPR treatments for the disease itself, however. Without an active Fanconi anemia pathway, cells may not be able to replace their mutated genes with normal genes after Cas9 makes a cut.
In fact, the level of activity of the Fanconi anemia pathway may affect how efficiently CRISPR can insert DNA in a specific cell. The researchers concluded that, while end-joining is the default repair mechanism after a double-strand break, the Fanconi anemia pathway competes with it, and that higher activity results in more homology-directed repair and less end-joining.
Cancer treatments
While the findings help scientists better understand the DNA repair mechanisms in human cells, they could also help researchers developing anti-cancer therapies that target DNA repair in cancer cells. Because other factors now appear to be involved in the repair of double-strand breaks, this research expands the list of proteins that could be misregulated in order to screw up DNA repair in cancer cells and make them more susceptible to death.
Richardson also found that one of the 21 proteins in the pathway, FANCD2, always homes in on the site of the double-strand break created by CRISPR-Cas9, indicating it plays an important role in regulating the insertion of new DNA into the genome at the cut site. FANCD2 could be tweaked to boost the frequency with which a cell inserts DNA via homology-directed repair.
“Also, since FANCD2 localizes to the site of Cas9 breaks, you can use FANCD2 to map where Cas9 is cutting in any cell type,” Richardson said. “If you edit a population of cells and you want to know where the on- and off-target cuts are, you can just map where FANCD2 was found in the genome and you can find the cuts.”
“The whole Fanconi anemia pathway affects the balance between end-joining and homology-directed repair; it acts like a traffic cop,” Corn said. “So a patient’s genotype will affect how you do gene editing.”
Other UC Berkeley authors are Katelynn Kazane, Sharon Feng, Elena Zelin, Nicholas Bray, Axel Schäfer and Stephen Floor, who is now at UCSF. The work was supported by grants from the Li Ka Shing Foundation, Heritage Medical Research Institute and Fanconi Anemia Research Foundation.
RELATED INFORMATION
- CRISPR-Cas9 genome editing in human cells works via the Fanconi Anemia pathway (Nature Genetics)
- Innovative Genomics Institute
- Jacob Corn’s lab website