Translating Genes’ Instructions
Sixty-three years ago, a drawing of a double helix appeared in a short scientific paper in the journal Nature. Since that milestone report by Watson and Crick, many thousands of researchers have sought to refine our understanding of the steps that lead from the intertwined strands of DNA to the assembly of proteins — the heavy lifters that make life possible.

What is known as “the central dogma of molecular biology” asserts that genes’ instructions, encoded in DNA, become transcribed into a related molecule, RNA, and ultimately translated to make proteins. But major questions remain about precisely how this fundamental feat is accomplished.
The Human Genome Project revealed that genes make up only a tiny fraction of the three billion bases, or coding “letters,” along human DNA strands. The other 98-plus percent are either relics of genetic code left over from ancient viral and jumping gene invasions, or they are instructions for regulating what would otherwise be a chaotic and error-prone gene copying process.
When a gene’s DNA sequence gets transcribed into RNA, the neighboring sequences that do not code for proteins are faithfully transcribed along with it. Somehow the gene must be plucked from its neighbors. UCSF biochemist Hiten Madhani studies the puzzling process by which gene and non-gene part company.
A molecular cutting machine called the spliceosome is known to snip bonds that link each gene’s sequence to its neighbors. Tens of thousands of spliceosomes can be at work in a cell at any given time.
Research suggests that the cuts are made at precisely the right location to separate gene from non-gene. “It’s all RNA — it all looks the same,” Madhani says. “How does the spliceosome select the exact site for cleavage?”
The answer matters. About ten percent of all cancers harbor mutations that disrupt the spliceosome’s precision performance, Madhani says. Mutations in the splicing machinery underlie most cases of a bone marrow cancer called myelodisplastic syndrome, for example.
Madhani’s lab has developed techniques to chemically tag and isolate spliceosomes in the act of cutting and splicing. He can determine precisely where they are cleaving the RNA.
He studies the process in a yeast called Cryptococcus neoformans, which would seem to have a rather stripped down splicing requirement. But each yeast cell has nearly 7,000 genes, along with some 40,000 non-coding sequences, called introns.
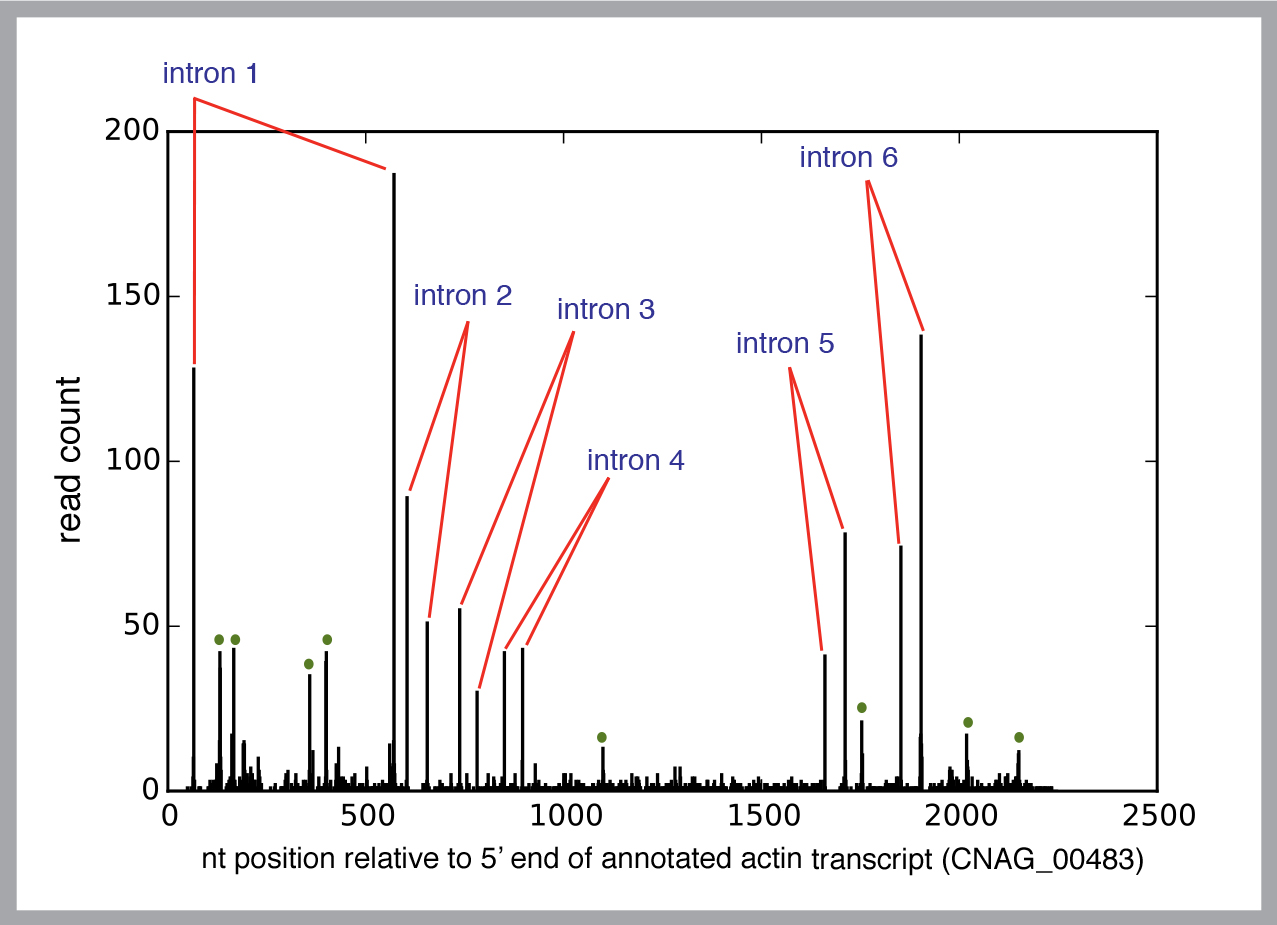
To gain a reliable picture of the process, Madhani needs to study a huge number of spliceosomes.
“Instead of looking at one intron from one gene at a time, we want to look at every intron in every gene. Since you’re not just looking at a single spliceosome, you can find basic principles.”
He employs “deep sequencing” technology, which allows automated analysis of all the spliceosomes active in a cell at a time. (This type of technology now makes it feasible to sequence the genome of a human for a few thousand dollars.) Each of his experiments can generate hundreds of millions of sequences to analyze. And therein lies a new challenge.
“We generate a lot of data — it’s very information-rich, and there is lots of potential for noise and artifacts that can keep you from being sure you are looking at what you think you’re looking at.”
Last year, as part of the Sackler Sabbatical Research Program, Madhani worked with Berkeley computational biologist Steven Brenner to tackle the mountains of data. He expects the research collaboration will help him sort through the possible explanations for spliceosomes’ almost uncanny precision.
“One hypothesis is that, in fact, the spliceosome makes a lot of mistakes,” he says. “How could it not? But we suspect that the cell employs a whole series of proofreading mechanisms that correct splicing errors.”
Almost every intron — 40,000 in yeast; more than 200,000 in humans — begins with same series of bases. “One way of proofreading could be to determine if that rule was followed or not,” he says.
Half a dozen hypotheses invite scrutiny. Without the ability to analyze millions of spliceosomes in action, Madhani says researchers would know only the outcome, not the process. “Like looking at the sausage instead of getting inside the sausage factory.”
Because spliceosome defects are linked to so many cancers, he thinks the research may lead to new treatments. Already, some biotech companies are pursuing ways to inhibit faulty spliceosome action, he says.
“I’m confident that if we can understand how abnormal RNA splicing contributes to cancer we will find ways to exploit this knowledge to get a step ahead of tumor growth.”
_________________________________
The Raymond and Beverly Sackler Center for Biomedical, Physical and Engineering Sciences rewards innovative research approaches that take advantage of and promote the convergence of the biomedical, physical and engineering fields, encouraging scientists to move into new fields and cross into other areas of convergence in order to realize the full potential for achieving transformative scientific breakthroughs. To advance this fertile research area, the Center facilitates innovation and faculty collaboration between UC Berkeley and UCSF through a prestigious sabbatical exchange program. For more information about the Sackler Center, please visit sackler.berkeley.edu.